
Varón de 29 años de edad derivado a consultas de cardiología para screening de miocardiopatía dilatada. Como único antecedente de interés, es fumador activo. Refiere encontrarse asintomático en el ámbito cardiovascular.
El caso índice es su padre, trasplantado cardiaco hace 3 meses por insuficiencia cardiaca avanzada secundaria a una miocardiopatía dilatada con disfunción ventricular izquierda severa y alta carga arrítmica. No se le realizó RMN en el único ingreso en nuestro centro, dado que su situación clínica no lo permitió, y se encuentra pendiente del resultado del estudio genético. El abuelo paterno de nuestro paciente falleció a edad temprana, desconociéndose la causa. Tiene una tía paterna, viva y que realiza seguimiento en otro centro por arritmias. El resto de familiares se encuentran aparentemente sanos y se están realizando también cribado.
Dentro del protocolo de screening de miocardiopatía dilatada, se realiza una analítica, sin hallazgos patológicos; un electrocardiograma, donde se aprecia fragmentación de la parte terminal del QRS en cara inferior, sin alteraciones de la repolarización; y un ecocardiograma. En esta prueba se describe un ventrículo izquierdo ligeramente dilatado, con función sistólica en el límite bajo de la normalidad (FEVI 52% por Simpson biplano) e hipoquinesia ligera de los segmentos basales y medios de septo inferior y cara inferior.
Ante los hallazgos del ecocardiograma y con la sospecha de una forma preclínica de miocardiopatía dilatada se amplía el estudio con TAC de arterias coronarias, que descarta la presencia de estenosis, y RMN cardiaca. Esta última confirma la dilatación del ventrículo izquierdo, que se gradúa como moderada, y la FEVI en el límite bajo de la normalidad (51%). Objetiva también una pequeña región disquinética en el segmento basal del VD, que por lo demás es funcional y estructuralmente normal. En las secuencias de realce tardío con gadolinio se objetiva una escara muy abigarrada y extensa, subepicárdica, que afecta de forma prácticamente circunferencial al ventrículo, respetando únicamente septo inferior y cara inferolateral.
Todos estos hallazgos son sugestivos de una variante patogénica en gen desmosómico. Por ello se solicita un Holter y una ergometría previo a la valoración en consulta de Cardiopatías Familiares. En el Holter se objetiva una taquicardia ventricular monomórfica no sostenida de 44 latidos, sintomática por palpitaciones. Se revisan los resultados del estudio genético de su padre, que resulta ser portador de una variante truncante en el gen DSP. Ante todos estos hallazgos, se discute el caso de nuestro paciente en sesión multidisciplinar, decidiéndose la indicación de DAI en prevención primaria.
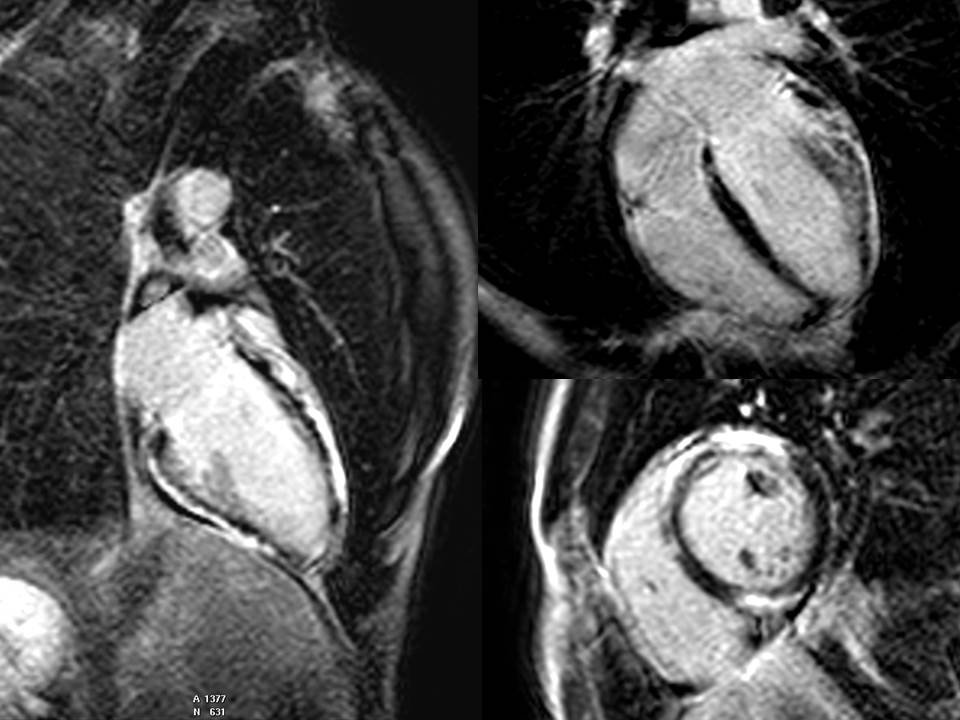
Imagen 1. Secuencia de realce tardío
Comentario
La miocardiopatía arritmogénica de ventrículo izquierdo (MAVI) es una entidad pobremente caracterizada, al contrario que ocurre con la de afectación predominantemente derecha, que consta de unos criterios diagnósticos definitivos. Los hallazgos ecocardiográficos de esta entidad son en ocasiones indistinguibles de formas incipientes de miocardiopatía dilatada familiar clásicas, como la secundaria a variantes patogénicas en TTN. Esto se debe a que aunque en ocasiones se aprecian alteraciones funcionales y/o estructurales en el ventrículo derecho, éstas pueden no estar presentes o incluso pueden pasar desapercibidas con esta técnica de imagen. De esta forma, pacientes que tienen un elevado riesgo de muerte súbita por su genotipo pueden estar desprotegidos durante meses hasta que se completa el estudio y se obtiene un resultado genético concluyente que disminuye el umbral para la indicación de DAI.
La resonancia magnética nuclear juega un papel fundamental en la detección precoz de estas formas de miocardiopatía arritmogénica. El artículo recientemente publicado por Augusto et al (Eur Heart J Cardiovasc Imaging, 2019) identifica algunas características por RMN cardiaca distintivas de esta entidad, como son la presencia de una escara subepipcárdica en anillo o la presencia de alteraciones de la contracción segmentaria de cualquiera de los dos ventrículos (siendo más específicas de MAVI las del ventrículo derecho). Pese a que nuestro paciente no cumplía criterios de miocardiopatía arritmogénica de VD y los hallazgos en ecocardiograma eran muy sutiles, presentaba ambos hallazgos en la RMN que se le realizó, lo que levantó las sospechas de una variante en un gen desmosómico. Esto aceleró el estudio y la estratificación del riesgo arrítmico y finalmente llevó de forma individualizada y fuera de las guías de práctica clínica actuales a la decisión de implante de DAI en prevención primaria, incluso antes de obtener el resultado del estudio genético en nuestro paciente. En resumen, los hallazgos de la RMN fueron determinantes de cara a evitar un eventual debut como muerte súbita.
Conclusión
Pese a la escasez de la evidencia disponible por no tratarse de las etiologías más frecuentes de miocardiopatía dilatada, las variantes en genes desmosómicos se han caracterizado por presentar un curso especialmente grave desde el punto de vista arrítmico, que no necesariamente va paralelo a la aparición de disfunción ventricular. Su identificación precoz, que en el futuro probablemente gire en torno a los hallazgos de la RMN y la generalización del estudio genético a todos los pacientes con MCD, permitirá establecer con mayor grado de fiabilidad el riesgo de estos pacientes en estadios precoces, así como valorar los criterios más adecuados para la indicación de implante de DAI en prevención primaria.
Ideas clave
- La resonancia magnética cardiaca es una herramienta fundamental para distinguir a los pacientes con miocardiopatía arritmogénica de ventrículo izquierdo frente a la miocardiopatía dilatada.
- El hallazgo de una escara subepicárdica en anillo por resonancia es muy específica de pacientes portadores de variantes patogénicas en genes desmosómicos.
Vídeo
Vídeo 1
- Ver vídeo. Imágenes en cine en todos los planos cardiacos
Autores
- Autores: Marta García Montero, Esther Pérez David, Mari Ángeles Espinosa, Raquel Prieto Arévalo, Ana González Mansilla, Candelas Pérez del Villar, Antonia Delgado, Elena Rodríguez, Javier Bermejo y Francisco Fernández Avilés. Hospital Universitario Gregorio Marañón (Madrid).
