





















































![La SEC te lleva a AHA24 [IN] ∙ Estudio SUMMIT](images/telleva/acc24/1.%20SUMMIT.png#joomlaImage://local-images/telleva/acc24/1. SUMMIT.png?width=600&height=336)
![La SEC te lleva a AHA24 [IN] ∙ Estudio BROOKLYN](images/filtros/cardiotv/25.%20BROOKLYN.jpg#joomlaImage://local-images/filtros/cardiotv/25. BROOKLYN.jpg?width=600&height=336)

![La SEC te lleva a AHA24 [IN] ∙ Estudio REALIZE-K](images/filtros/cardiotv/18.%20REALIZE-K.jpg#joomlaImage://local-images/filtros/cardiotv/18. REALIZE-K.jpg?width=600&height=336)
![La SEC te lleva a AHA24 [IN] ∙ Estudio SARAH](images/filtros/cardiotv/20.%20SARAH.jpg#joomlaImage://local-images/filtros/cardiotv/20. SARAH.jpg?width=600&height=336)
![La SEC te lleva a AHA24 [IN] ∙ Estudio CLEAR SYNERGY (OASIS9)](images/filtros/cardiotv/14.%20CLEAR%20OASIS9.jpg#joomlaImage://local-images/filtros/cardiotv/14. CLEAR OASIS9.jpg?width=598&height=336)



Paciente mujer de 37 años que es trasladada en 2005 desde otro centro tras presentar un cuadro de insuficiencia cardiaca aguda.
La paciente no presentaba ningún antecedente médico de interés salvo anemia ferropénica ya estudiada y relacionada con la menstruación. No refiere antecedentes familiares relevantes.
Manifiesta disnea de moderados esfuerzos de 2 meses de evolución y comienzo de tos una semana antes. No presentaba fiebre ni dolor torácico.
Tras 3 días de aparente infección respiratoria comenzó con disnea de mínimos esfuerzos y 2 días después desarrolló disnea paroxística nocturna.
La exploración física a su llegada a nuestro centro mostraba: presión arterial de 100/60 mmHg; FC: 140 lpm; saturación del 91% con O2 a 2 litros por minuto. La paciente está pálida y fría, con pulso venoso yugular aumentado, ritmo de galope por tercer tono y crepitantes bibasales ligeros. ECG: taquicardia sinusal a 140 lpm con bajo voltaje en derivaciones de miembros y mala progresión de R en cara anterior. PR normal. QRS 80 msg. La radiografía de tórax mostraba cardiomegalia y edema intersticial.
La analítica mostró: Hb 8,5 g/ dl (VCM:76) y 13800 leucocitos con 83% neutrófilos; presentaba elevación de ALT, AST, Amilasa y LDH con GGT, fosfatasa alcalina y bilirrubina normal. El Nt-proBNP era de 9929 pg/ml. El filtrado glomerular era normal (70 ml/min) la CK y la troponina I fueron normales. El ecocardiograma mostró un ventrículo izquierdo moderadamente dilatado, con hipoquinesia global y fracción de eyección biventricular muy deprimida (FEVI:18%), insuficiencia mitral moderada y un patrón restrictivo en el llenado mitral.
Con el diagnóstico de miocardiopatía dilatada idiopática (MCD) e insuficiencia cardiaca congestiva aguda ingresa en unidad coronaria donde se mantiene estable bajo tratamiento con diuréticos intravenosos y dosis altas de dopamina y dobutamina. Se realizó cateterismo derecho que objetivó: AD: 12 mmHg; VD: 44/12; PAPm:29; PCP: 25. El índice cardiaco era de 2,2 l/min/m2. Coronariografía sin lesiones. Se realizó biopsia endomiocárdica que no mostró signos de miocarditis.
Tras 4 días de estabilidad clínica en los que no se produce mejoría de la función ventricular sufre empeoramiento progresivo con hipotensión, reducción de diuresis y necesidad de mayores dosis de fármacos inotrópicos. Se inserta balón de contrapulsación pero el empeoramiento persiste por lo que el sexto día se implanta asistencia biventricular como puente al trasplante (TxC).
Tras un procedimiento sin incidencias es extubada el segundo día post-intervención y se retiran inotrópicos y balón de contrapulsación. No evidencia de mejoría ecocardiográfica de la función ventricular y tras 8 días de asistencia circulatoria se realiza TxC ortotópico. Veintiún días tras el TxC es dada de alta en buen estado.
En 2010 la paciente participa en un proyecto de investigación destinado a evaluar la prevalencia de mutaciones en genes desmosómicos en pacientes TxC por MCD. Se secuencian los genes JUP, PKP2, DSG2, DSC2, DSP encontrándose dos mutaciones en el gen DSP (R907H y R1838H) que no habían sido descritas con anterioridad. Ambas mutaciones no estaban presentes en 200 controles.
Se procedió a realizar un estudio familiar exhaustivo encontrando que la madre de la paciente de 69 años había sido diagnosticada de MCD con disfunción ligera (FEVI 48%) y fibrilación auricular 1 año después del TxC de su hija. Ella y una hermana de 40 años (sin signos de MCD) eran portadoras de ambas mutaciones mientras que las 2 hijas no las habían heredado.
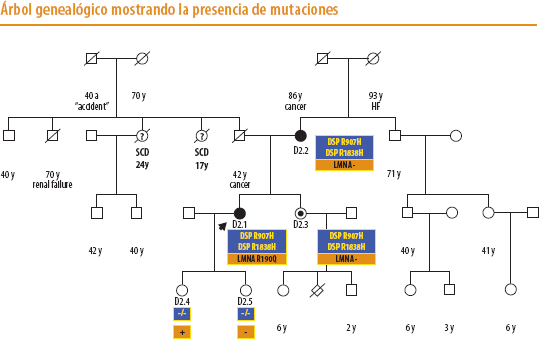
Sin embargo, llamaba la atención la gran discordancia en el curso clínico de la enfermedad entre la paciente índice y su madre/hermana y la existencia de 2 tías por parte paterna fallecidas por muerte súbita a los 24 y 17 años respectivamente. Se decidió analizar el gen LMNA en la paciente índice por su asociación a MCD y muerte súbita y se documentó una mutación ya descrita (R190Q).
La madre y hermana no presentaban la mutación en LMNA, lo que podría explicar el curso clínico más favorable de la enfermedad en ellas.
Los hallazgos genéticos permitieron proporcionar consejo clínico, reproductivo y profesional a otros miembros de la familia.
Referencias
1. Garcia-Pavia P, Syrris P, Salas C, Evans A, Mirelis JG, Cobo-Marcos M, et al. Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: A clinicopathological study. Heart 2011;97:1744-52.
2. van Rijsingen IA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol. 2012;59:493-500.
3. Hershberger RE, Siegfried JD. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2011;57:1641-9.
















.png)